熱線:021-66110819,13564362870
Email:info@vizai.cn
熱線:021-66110819,13564362870
Email:info@vizai.cn
光生物制氫效果很好 其重要性,因為它有望產生清潔的可再生能源。 在自然界中,綠藻不能產生氫氣,因為 氫化酶對氧氣極度敏感的結果。 然而,我們發現硅化誘導的綠藻 聚集體可以實現可持續的光生物氫 即使在自然有氧條件下也能生產。 核心- 綠藻聚集體的殼結構創造了一種平衡 光合電子產生和氫化酶之間 活性,從而允許產生氫氣。 這一發現 為太陽能驅動的水分解提供了可行的途徑 轉化為氫氣和氧氣以開發綠色能源替代品 通過使用合理設計的細胞-材料復合物。
氫氣(H2 ) 被認為是一個有前途的替代品 對于化石燃料,由于其卓越的轉換效率, 環保,高能量容量。 [1] H2 今天的生產主要依賴蒸汽 碳氫化合物重整、煤氣化和 核動力水電解,這是能源密集型和不可持續的。 [2] 太陽能生物制氫 電源提供了產生 H2 的可能性 那是可再生的 和碳中和,因為它直接使用取之不盡的資源:太陽能和來自 H2O 的電子。 [3] 在自然界, 光合微生物,特別是綠藻,可以 使用氫化酶(一種酶 催化分子氫的可逆氧化) 耦合到光合作用機器。 [4] 然而,這是 在幾分鐘內發生的短暫過程 暗光過渡。 [5] 這是因為氫化酶失去了它的 在有氧的情況下發揮作用。 [6] 在黑暗中,細胞呼吸產生激活氫化酶的厭氧條件。 [7] 在從黑暗走向光明的瞬間, 來自光系統 II (PSII) 反應中心的水光氧化反應 (H2O!2H+ + 1/2O2 + 2 e¢ ) 的光合電子可以傳遞給氫化酶以產生 H2 (2H+ + 2 e¢ !H2)。 然而 光合作用反應迅速產生氧氣 使氫化酶失活。 [8] 為了提高光生物 H2 產量,科學家篩選了 [9a,b] 和 構建了具有改善的氧耐受性的突變體和 通過降低內在氫化酶氧敏感性 各種策略。 [9c] 不幸的是,進展有限 制作。 目前,硫剝奪是最常見的 用于產生綠色厭氧環境的方法 藻類。 [7a] 然而,這種代謝處理可以同時抑制 PSII 活性并逐漸終止隨后的 H2 產生。 [7a, 10] 由于不利結果,綠藻在 H2 中的大規模應用 生產仍然不現實。
在自然界中,許多生物體,如硅藻, 球石和趨磁細菌已經從生物礦化過程中發展出特定的礦物結構 提供廣泛的保護和獨特的功能。 [11] 近期 研究成果表明,仿生礦化可以 作為一種有用的工具來改造細胞和病毒,以及 由此產生的細胞-材料復合物總是有不同的 來自原生生物的生物學特性。 [12] 這里我們 報告一種可以賦予綠藻的仿生硅化 具有可持續光生物 H2 的新能力 在自然大氣條件下生產。
蛋白核小球藻 (C. pyrenoidosa) 是一種單細胞 綠藻在其葉綠體中具有突出的核糖體, 也是少數商業化的微藻物種之一 已被大規模培養。 通常,細胞是 在單細胞狀態下,直徑 3-4 毫米,沒有傾向 自聚合(圖 1a)。 聚(二烯丙基二甲基氯化銨)(PDADMAC) 分子可以模擬硅化蛋白誘導 原位二氧化硅沉積在細胞表面, [12b, e] 這可以 也適用于綠藻。 仿生后 修改后,硅化的 C. pyrenoidosa 細胞可以自聚集(圖 1b)。 因此,硅化細胞具有 降低的 zeta 電位(約 ¢ 3 mV)與 原生細胞(約 ¢ 15 mV),這有利于 粒子團聚(支持信息,圖 S1)。 我們還確認細胞通過 無定形SiO2 (支持信息,圖S2)形成 細胞材料在硅化過程中聚集。 這 通過調節細胞硅化中的 C. pyrenoidosa 密度可以將聚集體尺寸控制在 10-500 mm 中(支持信息,圖 S3 和 S4)。

圖 1. 蛋白核小球藻細胞及其聚集體。 a) 本地人 使用光學顯微鏡和掃描電子觀察細胞 顯微鏡(SEM)。 比例尺:20 毫米(插圖:2 毫米)。 b) 聚合 使用光學顯微鏡和 SEM 觀察細胞。 比例尺: 50 毫米(插圖:2 毫米)。 c) 含有 WO3 的天然小球藻培養基 粉末(管的底部)。 d) 本地和聚合的小球藻培養物 管中裝有 WO3 粉末的介質。 e) 聚合小球藻 含有 WO3 粉末的培養基(管底)。 酒吧 表示 WO3 的標準顏色變化 在 H2 存在下 在 解決方案。
一些金屬氧化物的變色性質 它們的氧化態已被用于方便的光學 H2 檢測。 [13] 氧化鎢 (WO3) 是常用的,因為它 與 H2 反應 生產鎢青銅,其顏色由淺黃色變為藍灰色。 [13] 這里將 WO3 粉末添加到細胞培養物中作為 環境大氣下 H2 產量的指標。 這 天然和聚集的 C. pyrenoidosa(約 100 毫米)的 H2 生產能力如圖 1 c-e 所示。 在這 實驗中,C. pyrenoidosa(總細胞密度 培養基為 3.0 × 107 細胞mL¢ 1 ) 被照亮 在 100 mEm¢ 2 s¢ 1 的光強度下 12 小時。 在本地 小球藻,WO3 粉末顏色不變表示無 可檢測 H2 在系統中(圖 1c),與 綠藻不能產生 H2 的傳統觀點 在自然有氧條件下。 然而,在綜合 小球藻,WO3 粉末的顏色變成藍灰色 (圖 1 e),在溶液中產生 H2 之后 濃度約 10 mmolL¢ 1 (支持信息, 圖 S5)。 需要注意的是,培養基是 暴露在空氣中,因此產生的 H2 聚集的細胞發生在自然大氣條件下。
為了定量研究聚集的小球藻細胞中 H2 產生的特性,我們監測了 H2 和 O2 在使用氣體的密閉玻璃管的頂部空間中 色譜法(圖 2a)。 含有小球藻 (1.2 × 108 細胞mL¢ 1 ) 加入 60 mL 光照下的密封玻璃管(100 mEm¢ 2 s¢ 1 )。 為了 原生小球藻,H2 無法檢測到頂空 在任何測量點,頂部空間中的 O2 含量從最初的 21% 增加到 23%。 結果表明,天然小球藻細胞進行光合 O2 進化,但不產生 H2,這是由于 在有氧條件下使氫化酶失活。 有趣的是,觀察到 100 毫米的聚集體以約 0.35 mmolH2h¢ 1 (mg葉綠素)¢ 1 (圖 2a;支撐 信息,圖 S6)。 這個比率是自然界瞬時生物質燃料產量的 1.75 倍 (0.20 mmolH2h¢ 1 (mg 葉綠素)¢ 1 ).[14] 更重要的是, 這種 H2 的產生不僅限于 幾分鐘; 相反,這種生產被檢測到 至少 48 小時。 隨后,H2 的速率 生產開始減少(支持信息, 圖 S6),這可能是由于聚合受損 聚集體中細胞增殖產生的結構(支持信息,圖 S7)。 的生產 在聚合之前,H2 可以持續至少 60 小時 結構分解(支持信息,圖 S6 和 S7)。 與原生小球藻相比,我們注意到 O2 聚集體頂部空間的含量從 實驗期間為 21% 至 19%。 這種減少很可能 歸因于消耗 O2 分子的細胞呼吸 存在于封閉系統中。 在聚集的細胞中,O2 進化減少,因此 H2 產生 增加。 此外,在交替的時期 明暗(圖 2 b),我們發現聚集體僅 在光照期間產生 H2; 下未檢測到 H2 黑暗的條件。 這種明暗切換效應暗示著 H2 的產生與光合作用直接相關。
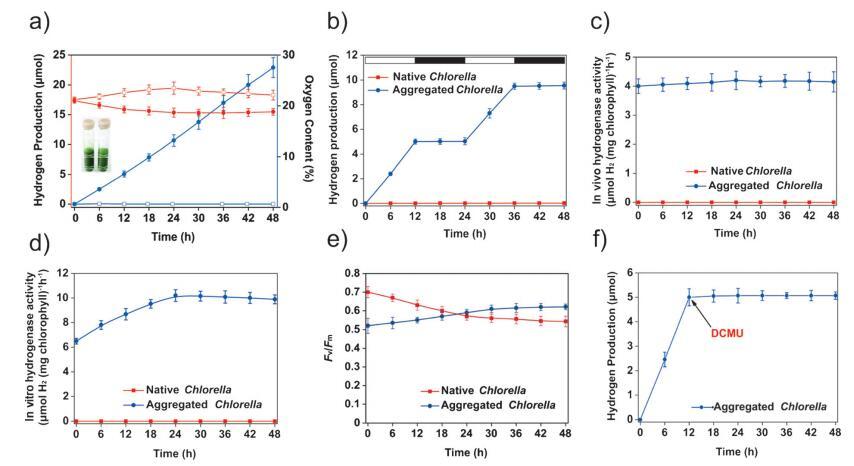
圖 2. 在有氧條件下聚集的小球藻光生物制氫。 a) H2的量和O2的含量 在 100 mEm¢ 2 s¢ 1 光強下密封管的頂部空間 在不同的時間段(n=6)。 紅線:O2%。 藍線:H2amount。 空心方塊:原生小球藻。 實心圓圈:聚集的小球藻。 b) 帶有聚集小球藻的密封管中的累積 H2 積累 在明暗過渡期間(n = 5)。 c) 天然和聚集的小球藻 (n=5) 中的體內氫化酶活性。 d) 體外氫化酶活性 天然和聚集的小球藻 (n=5)。 e) 光強度下天然和聚集的小球藻 PSII (Fv/Fm) 的最大量子產率 100 mEm¢ 2 s¢ 1 在不同的時間段(n=3)。 f) DCMU 對聚集的小球藻 (n=3) 產生的 H2 的影響。
在綠藻中,光合水分解在功能上與活化的氫化酶產生的 H2 相關。 在 天然小球藻,既不是體內也不是體外氫化酶 可以檢測到活性(圖 2c 和 d),因為氫化酶在環境存在的情況下失活 氧氣。 [6] 在小球藻聚集體中,體內氫化酶 活動在前 24 小時內略有增加,然后保持不變 在 4 mmolH2h¢ 1 的相對穩定水平 (mg葉綠素)¢ 1 (圖 2 c),這也高于 H2 的速率 從大氣中生產氬氣(支持信息)。 該結果表明,氫化酶在 聚集體在大氣氧水平下仍然活躍。 體外檢查證實存在氫化酶 聚合中的活動,并且值甚至從 6.5 至 10.3 mmolH2h¢ 1 (mg 葉綠素)¢ 1 在前 24 小時內 (圖2d)。 這些結果表明聚合 處理可能有利于氫化酶的表達 提高 H2 生產潛力。
除氫化酶外,影響持續光生物 H2 產生的另一個重要因素是其催化作用。 底物,即光合電子,起源于 來自 PSII 反應中的水氧化反應 中心。[15] PSII 是一種大的多亞基膜蛋白 可以捕獲光子能量、分離電荷和 驅動類囊體膜中的電子轉移。 [16] 該 PSII 的最大量子產率 (Fv/Fm) 已被廣泛研究 用于評估 PSII 的活性。 Fv/Fm 的值在 實驗開始時原生小球藻為 0.7, 并最終降至約 0.5(圖 2e)。 這個 減少很可能是密封環境引起的應力的結果。 聚合中的 Fv/Fm 參數 小球藻在開始時減少到大約 0.5 實驗(圖 2 e),可能是由聚合處理引起的。 然而,最大量子產率 聚集的小球藻中的 PSII 從最初的 值 0.52 到最終值 0.62(圖 2e)。 結果 表明適當的聚集處理幾乎 不影響封閉細胞中 PSII 的活性,不像 硫剝奪治療的影響。 [17] 化合物 3-(3,4-二氯苯基)-1,1-二甲基脲 (DCMU) 是 一種特定的 PSII 抑制劑,可破壞 從 QA 到 QB 的電子, [17] 及其添加到聚合 即使在光照條件下也完全終止制氫 (圖2f)。 這一現象證實了 氫化酶與光合作用機器之間的耦合以產生 H2 在聚集的細胞中。
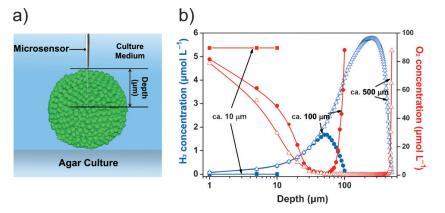
圖 3. 聚集小球藻的空間功能分化。 a) 微傳感器測量圖。 b) 氫氣 (藍色)和 O2 (紅色的) 不同大小的聚集小球藻的微觀結構 (n=5)。
微傳感器是研究生物的可靠工具 聚集體中的微環境(圖 3a;支持信息,圖 S8)。 在這里,我們使用 H2 和 O2 微型傳感器來檢查我們的聚集體中的內部環境。 在 100 毫米的骨料中,H2 的產量不 在骨料表面檢測到。 H2濃度 隨著探頭深度的增加而增加,并達到 最大值(約 1.7 mmolL¢ 1 ) 在聚合核心 (圖 3 b)。 此配置文件暗示 H2 是由核心產生的 細胞而不是表面細胞。 相比之下,O2 濃度隨著探針深度的增加而降低。 這 核心微環境缺氧,O2 核心中心的濃度幾乎為零,其中 氫化酶被激活,從而產生 H2。 聚合的空間 H2 和 O2 分布 小球藻細胞暗示存在空間功能 細胞之間的分化(SFD)。
聚合可以被認為是自生的 具有分化細胞功能的結構化核殼復合體。 表面細胞暴露在開放環境中,其功能與原生細胞相似 藻類。 然而,外面的細胞也起到了外殼的作用 防止環境 O2 滲透 進入聚合 核。 內細胞的呼吸反應消耗了任何 擴散的 O2 或光合作用產生的 O2 創造 缺氧域。 這種厭氧條件是由于 O2 擴散、PSII 產生的 O2 和隔離系統中的細胞呼吸之間的動態平衡由下式定義 封閉的外殼空間。 氫化酶和 PSII 活性 可以在這個域中保持,并且它們的耦合 保證可持續的光生物 H2 生產。 這 從個體綠藻到功能轉變 聚集的綠藻可以通過 SFD 來理解 核殼結構:核心細胞產生氫氣 由于隔離的厭氧微環境而產生的能力; 殼細胞保留了原生藻類的功能和 將核心細胞與開放環境隔離 (方案1)。
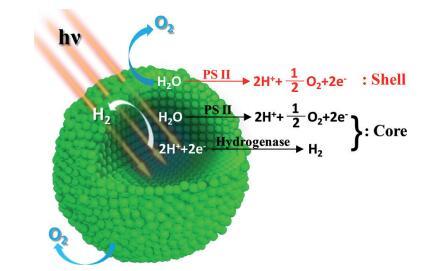
方案 1. 聚集小球藻的空間功能分化 細胞。
基于核殼的 SFD 對容量的影響 可以通過控制聚集體的大小來調整光生物 H2 的產生。 對于具有 直徑約 10 毫米,H2 濃度在兩個 可以忽略內部和外部單元格(圖 3 b)。 O2 濃度保持在同一水平(約 90 mmolL¢ 1) 從外部到核心(圖 3 b)。 結果表明 直徑為 10 毫米。 然而,隨著骨料尺寸的增加,更多的核心 細胞被封閉并與開放環境隔離。 微傳感器測量表明大聚集體 (500 mm) 表現出比 100 毫米骨料。 H2 濃度達到 5.3 mmolL¢ 1 在大集合的核心,而 O2 隨著探頭深度的增加,濃度降至零 增加到近 50 毫米(圖 3 b)。 因此,一個最優 用于光生物 H2 生產的 SFD 高度依賴 在總規模上。
在密閉玻璃管中的實驗表明, 總體而言,相對于 10 毫米和 500 毫米的骨料, 100 mm 聚集體(具有相同的初始總葉綠素 content) 表現出卓越的制氫能力 (支持信息,圖 S9 和圖 S10)。 這 10 毫米的骨料幾乎不能產生 H2 或性能 氫化酶活性。 頂空 H2 的量 由 500 毫米骨料產生的少于那些 由 100 毫米骨料在不同時間段產生的 (支持信息,圖 S9)。 雖然體外 500 mm 聚集體中的氫化酶活性較高, 500 mm 聚集體的體內氫化酶活性低于 100 mm 聚集體(圖 2 c 和支持信息,圖 S11)。 這很可能 由于內部光合作用的不利限制 PSII 產生電子,這證明了 測量 PSII 活動(支持信息,圖 S12)。 這表明氫化酶和 PSII 是保證光生物 H2 生產的關鍵。 PSII 效率的檢查表明 Fv/Fm 500 毫米聚集體的值僅為 0.1-0.3。 沒有 充足的光合電子,大細胞中的核心細胞 聚合體不能通過氫化酶有效地產生 H2 盡管氫化酶的活性很高。
眾所周知,利用有氧光合作用 在有氧條件下能產生H2的微生物 將是生物 H2 向前邁出的重要一步 生產。 [18] 然而,篩選仍然是一個巨大的挑戰 對于具有顯著氧耐受性的天然物種。 最近,黃等人。 鑒定了一種新的微藻菌株 (Chlorella vulgaris YSL01) 并且該菌株可以連續 產生高達 1.9 mLH2 (Lculture)¢ 1 與 5% O2 在 10% 二氧化碳。 [9b] 然而,這種 H2 的產生只發生在 人工的而不是自然的有氧環境。 在我們的 研究表明,聚集的綠藻(C. pyrenoidosa)可以 連續產生約 0.5 mLH2 (30 mLculture)¢ 1, 相當于約 17 mLH2 (Lculture)¢ 1 . 這個 自然條件下的價值約為 9 倍以上 改良條件下的普通小球藻 YSL01。 [9b] 我們的 研究表明,綠藻聚集體是 在曝光期間能夠光合自養 H2 到連續照明,這提供了第一種情況 在自然有氧條件下持續產生光生物 H2。 這一成就對于促進 綠色能源發展。
在我們的嘗試中,原位硅化可以誘導細胞 C. Pyrenoidosa 的聚集,因為細胞可以 由二氧化硅材料凝聚,這取決于細胞表面的仿生化學修飾。 這種化學品—— 材料細胞工程可以產生H2的新功能 通過新型細胞材料復合物生產 可行、廉價且有效。 更一般地說, 類似的基于化學材料的細胞修飾可能 擴展到其他微生物以誘導設計的 功能轉換。 它進一步遵循生物礦化啟發的策略,通過化學和 物質途徑。
這項研究得到了基礎研究的支持 中央高校資助項目(浙大校長項目) 和國家自然科學基金 (21471129 和 31370270)
[1] a) O. Kruse, B. Hankamer, Curr. Opin. Biotechnol. 2010, 21, 238 – 243; b) K. Srirangan, M. E. Pyne, C. P. Chou, Bioresour. Technol. 2011, 102, 8589 – 8604.
[2] a) J. A. Turner, Science 1999, 285, 687 – 689; b) J. R. Bartels, M. B. Pate, N. K. Olson, Int. J. Hydrogen Energy 2010, 35, 8371 – 8384.
[3] S. J. Burgess, B. Tamburic, F. Zemichael, K. Hellgardt, P. J. Nixon, Adv. Appl. Microbiol. 2011, 75, 71 – 110.
[4] a) A. Hemschemeier, T. Happe, Biochim. Biophys. Acta Bioenerg. 2011, 1807, 919 – 926; b) S. Grewe, M. Ballottari, M. Alcocer, C. D?Andrea, O. Blifernez-Klassen, B. Hankamer, J. H. Mussgnug, R. Bassi, O. Kruse, Plant Cell 2014, 26, 1598 – 1611.
[5] H. Gaffron, J. Rubin, J. Gen. Physiol. 1942, 26, 219 – 240.
[6] S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong, T. Happe, Proc. Natl. Acad. Sci. USA 2009, 106, 17331 – 17336.
[7] a) A. Melis, L. Zhang, M. Forestier, M. L. Ghirardi, M. Seibert, Plant Physiol. 2000, 122, 127 – 133; b) M. L. Ghirardi, L. Zhang, J. W. Lee, T. Flynn, M. Seibert, E. Greenbaum, A. Melis, Trends Biotechnol. 2000, 18, 506 – 511.
[8] E. Eroglu, A. Melis, Bioresour. Technol. 2011, 102, 8403 – 8413.
[9] a) A. Bandyopadhyay, J. St?ckel, H. Min, L. A. Sherman, H. B. Pakrasi, Nat. Commun. 2010, 1, 139; b) J.-H. Hwang, H. C. Kim, J.-A. Choi, R. A. I. A. -Shanab, B. A. Dempsey, J. M. Regan, J. R. Kim, H. Song, I.-H. Nam, S.-N. Kim, W. Lee, D. Park, Y. Kim, J. Choi, M.-K. Ji, W. Jung, B. H. Jeon, Nat. Commun. 2014, 5, 3234; c) M. L. Ghirardi, J. Cohen, P. King, K. Schulten, K. Kim, M. Seibert, Proc. SPIE-Int. Soc. Opt. Eng. 2006, 6340, U257 – U262.
[10] a) T. K. Antal, T. E. Krendeleva, T. V. Laurinavichene, V. V. Makarova, M. L. Ghirardi, A. B. Rubin, A. A. Tsygankov, M. Seibert, Biochim. Biophys. Acta Bioenerg. 2003, 1607, 153 – 160; b) A. A. Volgusheva, V. E. Zagidullin, T. K. Antal, B. N. Korvatovsky, T. E. Krendeleva, V. Z. Paschenko, A. B. Rubin, Biochim. Biophys. Acta Bioenerg. 2007, 1767, 559 – 564.
[11] a) C. E. Hamm, R. Merkel, O. Springer, P. Jurkojc, C. Maier, K. Prechtel, V. Smetacek, Nature 2003, 421, 841 – 843; b) F. Nudelman, N. A. J. M. Sommerdijk, Angew. Chem. Int. Ed. 2012, 51, 6582 – 6596; Angew. Chem. 2012, 124, 6686 – 6700; c) E. B?uerlein, Angew. Chem. Int. Ed. 2003, 42, 614 – 641; Angew. Chem. 2003, 115, 636 – 664.
[12] a) B. Wang, P. Liu, W. Jiang, H. Pan, X. Xu, R. Tang, Angew. Chem. Int. Ed. 2008, 47, 3560 – 3564; Angew. Chem. 2008, 120, 3616 – 3620; b) S. H. Yang, K.-B. Lee, B. Kong, J.-H. Kim, H.-S. Kim, I. S. Choi, Angew. Chem. Int. Ed. 2009, 48, 9160 – 9163; Angew. Chem. 2009, 121, 9324 – 9327; c) E. H. Ko, Y. Yoon, J. H. Park, S. H. Yang, D. Hong, K.-B. Lee, H. K. Shon, T. G. Lee, I. S. Choi, Angew. Chem. Int. Ed. 2013, 52, 12279 – 12282; Angew. Chem. 2013, 125, 12505 – 12508; d) G. Wang, X. Li, L. Mo, Z. Song, W. Chen, Y. Deng, H. Zhao, E. Qin, C. Qin, R. Tang, Angew. Chem. Int. Ed. 2012, 51, 10576 – 10579; Angew. Chem. 2012, 124, 10728 – 10731; e) W. Xiong, Z. Yang, H. Zhai, G. Wang, X. Xu, W. Ma, R. Tang, Chem. Commun. 2013, 49, 7525 – 7527.
[13] T. H?bert, L. Boon-Brett, G. Black, U. Banach, Sens. Actuators B 2011, 157, 329 – 352.
[14] L. J. Iwuchukwu, M. Vaughn, N. Myers, H. O?Neill, P. Frymier, B. D. Bruce, Nat. Nanotechnol. 2009, 5, 73 – 79.
[15] M. L. Ghirardi, S. Kosourov, A. Tsygankov, M. Seiber-t, Proceedings of the 2000 DOE Hydrogen Program Review (San Ramon, CA) 2000, pp. 1 – 13. NREL/CP-570-28890.
[16] F. Rappaport, B. A. Diner, Coord. Chem. Rev. 2008, 252, 259 – 272.
[17] A. Volgusheva, S. Styring, F. Mamedov, Proc. Natl. Acad. Sci. USA 2013, 110, 7223 – 7228.
[18] G. Goldet, A. F. Wait, J. A. Cracknell, K. A. Vincent, M. Ludwig, O. Lenz, B. Friedrich, F. A. Armstrong, J. Am. Chem. Soc. 2008, 130, 11106 – 11113.
 相關新聞
相關新聞